Seq2Pocket is a framework for protein ligand binding site (LBS) prediction that maps sequence-level predictions to 3D structural pockets. The method utilizes finetuned protein language model (pLM) to identify binding residues and restores pocket continuity through a two-step structural refinement process:
-
Embedding-Supported Smoothing: An additional classifier leverages latent embeddings of neighboring residues to fill in gaps and resolve spatial incompletion inherent in independent residue-wise predictions.
-
Structure-based Clustering: A surface-based clustering that utilizes Solvent Accessible Surface (SAS) points to group refined predictions into distinct, biologically relevant 3D pockets.
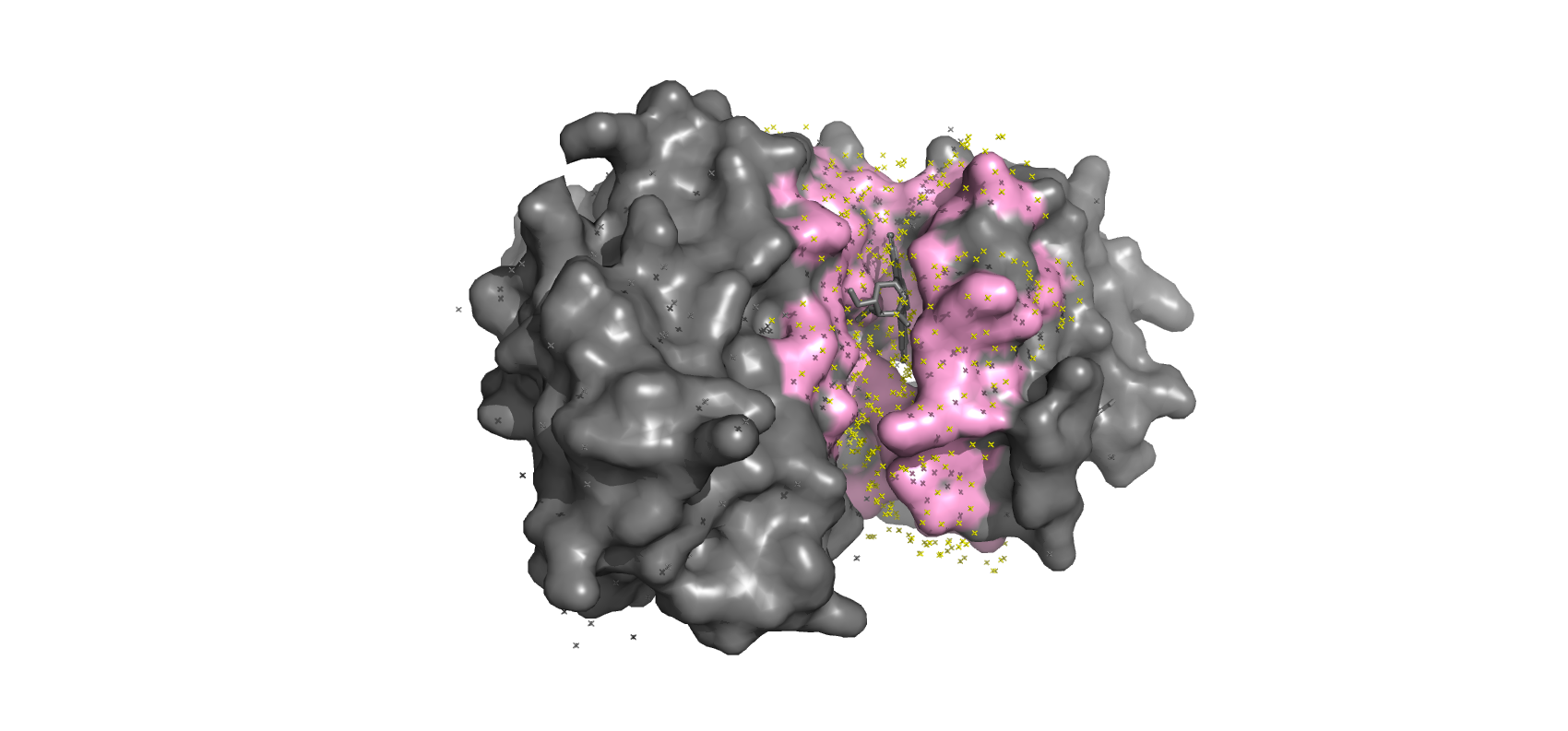
The Seq2Pocket Pipeline. Our framework takes residue-level probabilities from a finetuned pLM, applies an embedding-supported smoothing classifier to restore pocket continuity, and utilizes a surface-based clustering to define final 3D binding regions. Here, the finetuned pLM correctly identifies the Staurosporine inhibitor binding site on the human C-terminal Src kinase (PDB ID = 1bygA).
You can check out the model for cryptic binding site prediction using on this website: https://cryptoshow.cz/
Refer to the /tutorial folder for running the model locally.
The Seq2Pocket workflow is divided into three main stages:
- pLM Fine-tuning: Scripts for fine-tuning ESM2-3B on ligand binding tasks.
- Smoothing Classifier: A module that uses latent embeddings of a residue and its neighbors to decide if additional residues should be included in a pocket, improving structural completeness.
- SAS Clustering: An optimized clustering approach using Solvent Accessible Surface (SAS) points to group predicted residues into distinct 3D pockets.
The source code is located in the src/ folder, which contains the following subfolders:
data-extraction: Scripts for processing dataset files.pLM-training: Training scripts for finetuning the ESM2-3B models for General Binding Site (GBS) prediction.smoothing-classifier: Implementation of the embedding-supported smoothing classifier, including training logic.evaluation: Benchmarking scripts that utilize out-of-the-boxscikit-learnclustering algorithms (e.g., DBSCAN, Mean Shift).clustering: Implementation of the proposed clustering methodology: MeanShift applied to Solvent Accessible Surface (SAS) points.stats: Calculation of data and figures for the manuscript.visualizations: Scripts for visualizing pockets in pyMOL.
The enhancement of the sc-PDB dataset is implemented in separate branch named 'scPDB_enhancement' in the CryptoBench repository.
To run the scripts, it might be useful to install the packages specified in requirements.txt.
To generate SAS points, update your biopython's SASA.py with the updated version from this repository (SASA.py).
- scPDB-Enhanced: Zenodo repository
- An updated version of the scPDB dataset containing experimentally observed binding sites and ions omitted in the original release.
- Data and model weights: Storage link
If you have any questions regarding the usage of the framework, comparing your method against the benchmark, or if you have any suggestions, please feel free to contact us by raising an issue!
If you find our work useful, please cite the paper:
- Vít Škrhák, Lukáš Polák, Marian Novotný, and David Hoksza. 2026. Seq2Pocket: Augmenting protein language models for spatially consistent binding site prediction. bioRxiv. https://doi.org/10.64898/2026.01.28.702257
or, if you prefer the BibTeX format:
@article {2026.01.28.702257,
author = {Škrhák, Vít and Polák, Lukáš and Novotný, Marian and Hoksza, David},
title = {Seq2Pocket: Augmenting protein language models for spatially consistent binding site prediction},
elocation-id = {2026.01.28.702257},
year = {2026},
doi = {10.64898/2026.01.28.702257},
publisher = {Cold Spring Harbor Laboratory},
abstract = {Protein-ligand binding site prediction (LBS) is important for many domains including computational drug discovery, where, as in other tasks, protein language models (pLMs) have shown a great promise. In their application to LBS, the pLM classifies each amino acid as binding or not. Subsequently, for the purposes of downstream analysis, these predictions are mapped onto the structure, forming structure-continuous pockets. However, their residue-oriented nature often results in spatially fragmented predictions. We present a comprehensive framework (Seq2Pocket) that addresses this by combining finetuned pLM with an embedding-supported smoothing classifier and an optimized clustering strategy. While finetuning on our enhanced scPDB dataset yields state-of-the-art results, outperforming existing predictors by up to 11\% in DCC recall, the smoothing classifier restores pocket continuity. Next, we introduce the Pocket Fragmentation Index (PFI) and use it to select a clustering approach that preserves a consistent mapping between predictions and ground-truth pockets. Validated on the LIGYSIS and CryptoBench benchmarks, our approach ensures that pLM-based predictions are not only statistically accurate but also useful for downstream drug discovery, while maintaining state-of-the-art performance.Competing Interest StatementThe authors have declared no competing interest.},
URL = {https://www.biorxiv.org/content/early/2026/01/31/2026.01.28.702257},
eprint = {https://www.biorxiv.org/content/early/2026/01/31/2026.01.28.702257.full.pdf},
journal = {bioRxiv}
}
This source code is licensed under the MIT license.